Doudna and Charpentier were the principal investigators in the labs that published their paper detailing CRISPR’s adaptability to gene editing in Science way back in 2012. Their work drove the way for a new frontier in genome engineering, revolutionising our perspective on gene editing once and for all. It is only befitting that we take a look at the way they review the impact that CRISPR-Cas9 had on the field.
Gene editing is not a new and certainly not a novel concept. It has been around since the late ‘70s, when gene replacement was first achieved in yeast. There were many discoveries along the way, including and not limited to the discovery of Zinc Finger Proteins, the discovery of Homology Directed Repair and non-homologous end joining, all new methods and techniques that slightly furthered the case that we might be able to, one day, edit the human genome. Then, in the 1990’s, scientists across the world managed to, for the first time, sequence the entire human genomes. Everyone thought that this would change the way we deal with human genetics forever. But none of these discoveries had the impact they’d been advertised to have, until now, that is.
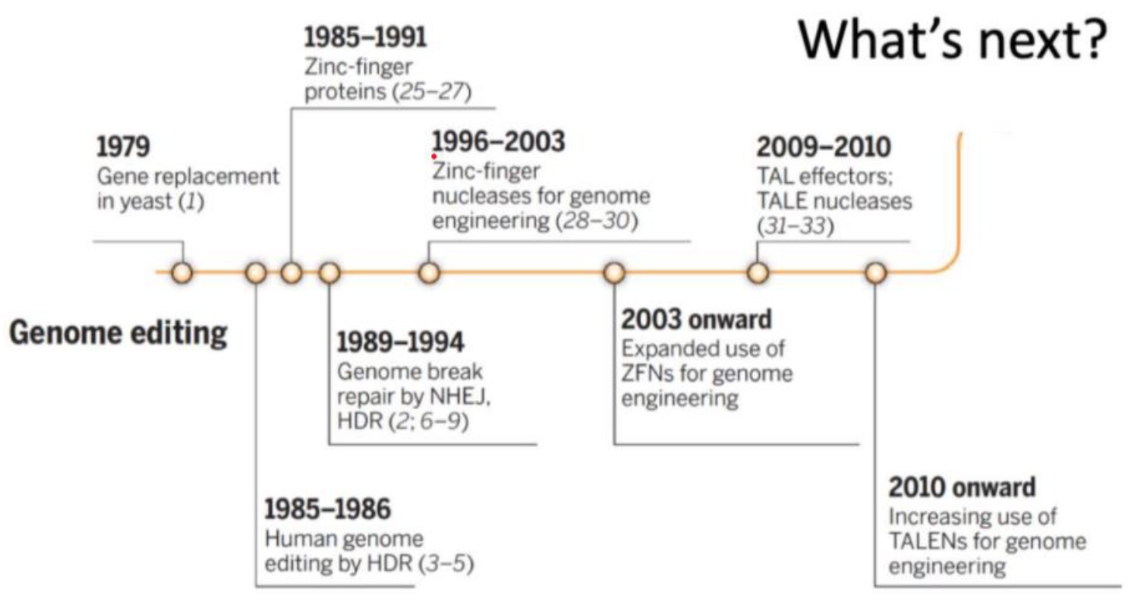
Fig 1. The timeline of progress on genome editing (without CRISPR).1
History of CRISPR-Cas
In the 2000’s, a range of laboratories began investigating CRISPR for unrelated reasons. They were found in certain bacteria and archaea, which seemed to be clustered repeats of certain genetic sequences. It was later discovered that the sequences seemed viral in origin or resembled certain plasmids. Further down the line, it was proposed that CRISPR-Cas was an adaptive immune response to past invasions, and that the sequences were antisense reminders of invading sequences. The same was established to be true in Streptococcus thermophilus. After this was it first hypothesised that bacteria could be weaponized to act as a deterrent to phages. This was tested successfully in 2008, and CRISPR RNA’s were shown to act as guides to locations that had to be spliced from invading genomes.
CRISPR-Cas: How does it work?
The operation of the CRISPR-Cas system is simple. Let us look into some basics on DNA first. DNA is a polymer comprised of four molecular bases, commonly represented as A, G, T and C. The structure of DNA is based on base-pairing, where A and T pair with each other and G and C pair do likewise. It is this pairing that results in a double stranded structure of DNA, where each strand perfectly binds with the opposing strand4. RNA has a structure that is similar to DNA, except for the T being replaced with a U. RNA is transcribed from DNA, and the RNA obtained is almost identical to the primary or the Watson strand of DNA, named after one of the researchers who discovered the structure of DNA. When the RNA is exported out of the nucleus, it is translated into proteins, which drive functions around the body. RNA, in and of itself, has the ability to base pair with a strand of DNA. RNA exists in nature usually as a single stranded molecule.
Fig. 2. DNA Base binding and complementarity.
In natural systems, invading virus/plasmid’s RNA have been reverse transcribed or reversed into DNA and subsequently integrated into host genomes in the form of repeated sequences. Hence, CRISPR stands for Clustered Regularly Interspaced Short Palindromic Repeats. All such sequences are clustered in a region in the host genome called the CRISPR locus. The Cas protein, prepared by the cellular machinery, and the RNA similar to the viral RNA, that was prepared by the host, form a complex and begin surveillance of the cytoplasm. This is known as the CRISPR-Cas surveillance complex.
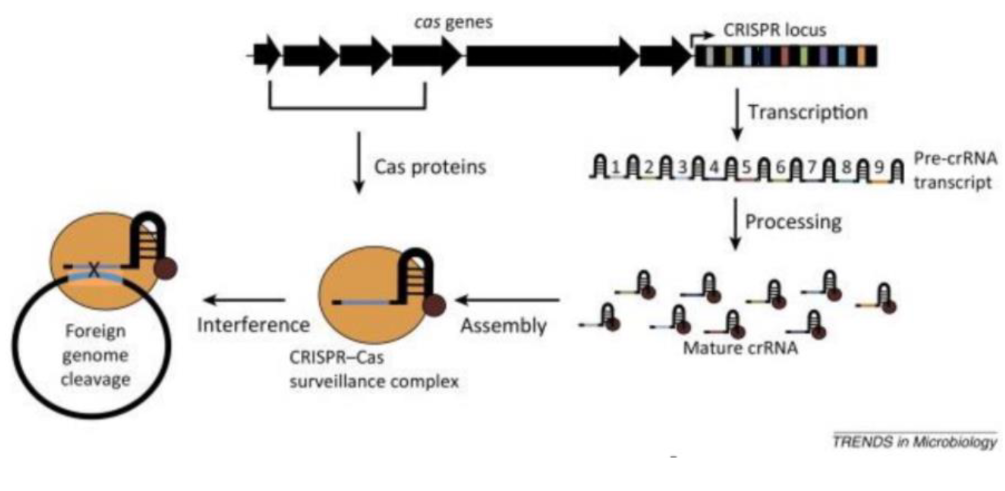
Figure 3. CRISPR Cas in Nature.5
The surveillance complex, now containing the CRISPR RNA’s, surveils the cell and uses the RNA to home to the sequence with which it shares complementarity. Once it homes in, the CRISPR-Cas system scans for a motif called a PAM motif, which must be present at the target of interest. If the PAM motif is found, the CRISPR-Cas complex binds to the DNA and induces what’s called a double stranded break (DSB). Once the break has been created, the cellular machinery of the invading pathogen stitches the broken area up, but without the sequence that had been cut. If the sequence coded for a protein that increased the pathogenicity of the invader, the invader is now disabled.
CRISPR-Cas: What can scientists do with it?
This very process can be exploited by scientists to edit the gene of their choice. The CRISPR RNA is around 23-55 nucleotides, and homes in to a sequence with perfect complementarity to induce a DSB. If the guide CRISPR RNA is engineered to reach the gene of interest, a DSB can be artificially induced anywhere. Techniques such as homology directed repair (HDR) also allow the cell to introduce a new sequence of interest at the location of a DSB. So, to modify sequence A into sequence B, the CRISPR-Cas system must be introduced into a cell, with the RNA sharing complementarity with A. Once the break is introduced, a HDR is initiated with sequence B, to make sequence B take A’s place in the genome. If the intent is to simply delete a gene, sometimes referred to as a knock-out, an alternate pathway referred to as non- homologous end joining is utilised. However, it is important to note that the sequences that are cut are usually very small, and the process must be tweaked to remove entire genes.
There are other ways CRISPR-Cas can be exploited. Since they recognise highly specific regions on the genome, they can also be used to map the structure of chromosomes in the relaxed state. They can also be used to make epigenetic modifications, modifications made directly to the genome by adding a structure to inhibit/ induce the gene.

Fig. 4: What to do after a DSB is induced.
The advantage that CRISPR has over all its predecessors is specificity and accuracy. The Cas9 protein will not bind to any region that doesn’t have both perfect complementarity to the RNA sequence it holds and the PAM motif mentioned above. While the latter might seem like a disadvantage, scientists seem to be on the verge of developing workarounds to that limitation.
Where do we go from here?
CRISPR-Cas9 has been revolutionary in academia and the impact has already started to extend to the clinical world. Human trials to treat genetic disorders are underway, with one woman in the United States being treated of sickle-cell anaemia using CRISPR therapy. While clinical applications progress, CRISPR has also given researchers vast potential to understand complex genes across various organisms and a quasi-regulatory mechanism, which can be exploited to understand the inner workings of various organisms. CRISPR is, hence, an invaluable tool in the toolkit of the microbiologist, the molecular biologist and the geneticist and most anyone trying to understand life.
But as we go down this path utilising one of the most powerful tools ever innovated, it is also the pertinent that we have discussions in the public forum on the ethics of different kinds of gene editing, and how legislation would be framed in this regard. A review of the legality worldwide, conducted in 2016, characterised large portions of genome editing policy worldwide as ‘vague’ and not sufficiently comprehensive. To truly utilise these amazing tools, we must make sure we account for the deleterious effects they could have when placed in the wrong hands, and ensure, to the greatest extent, that the tools are not misused as we move forward into a new frontier of human history.
Written by
Reference
Doudna, J. A. & Charpentier, E. The new frontier of genome engineering with CRISPR- Cas9. Science 346, (2014). doi: 10.1126/science.1258096.